Genetik von Herz-Kreislauf-Erkrankungen
Einleitung
Monogene Erkrankungen
Publikationen
Komplexe Erkrankungen
Publikationen
Angewandte Methoden
Sitz der Ag
Ständige Mitglieder
Unser Hauptinteresse gilt der Identifizierung neuer Gene, die eine entscheidende Rolle im komplexen Geschehen der Blutdruckregulation,
des Fettstoffwechsels und bei der Entstehung von Herz-Kreislauf-Erkrankungen spielen. Unsere Forschung richtet sich auf die Aufklärung
monogener und komplexer genetischer Erkrankungen. Monogene Syndrome erlauben die Genkartierung und Klonierung einzelner Gene, die für die Ausprägung des entsprechenden Phänotyps zuständig sind. Genetische Untersuchungen komplexer Erkrankungen können sowohl auf Kopplungsanalysen als auch auf Assoziationsstudien basieren. Kopplungsanalysen ermöglichen die Identifizierung von neuen, noch unbekannten Genorten (wo ist es), während Assoziationsstudien die Testung von Varianten in Kandidatengenen erlauben (was ist es). Wir haben ein umfangreiches Forschungsprogramm initiiert, um die Genetik von Herz-Kreislauf-Erkrankungen zu beleuchten.

Monogene Erkrankungen
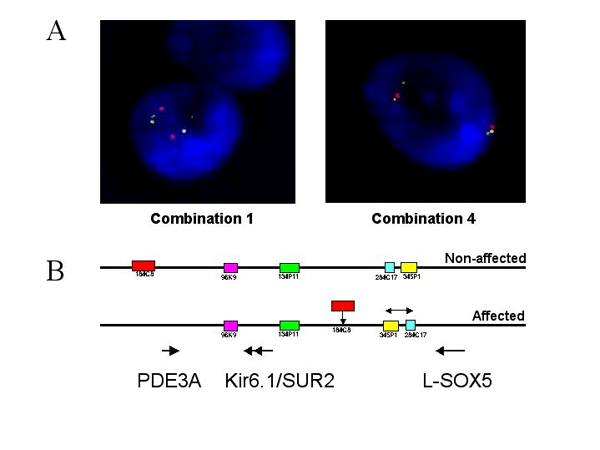
Unser Kooperationspartner, Prof. Nihat Bilginturan, beschrieb im Jahre 1973 eine im Norden der Türkei lebende Familie mit autosomal-dominant vererbtem Bluthochdruck und Kurzfingrigkeit (Brachydaktytlie). Im Jahre 1994 untersuchten wir diese Familie erneut und kartierten das verantwortliche Gen auf den kurzen Arm von Chromosom 12. Seitdem haben wir zusätzliche Familien mit dem Syndrom aus Frankreich, Kanada, den USA und Südafrika rekrutiert. Die Hypertonie wurde durch uns umfassend charakterisiert und es zeigte sich, dass der Blutdruck unabhängig von der Salzaufnahme ist. Dafür konnten bei alle betroffenen Personen Schlingen der posterioren inferioren cerebellären Arterie (sog. PICA) nachgewiesen werden, die einen neurovaskulären Kontakt zur ventrolateralen Medulla, einem Bereich des Hirnstamms, der für die Regulation des autonomen Nervensystems wichtig ist, vermitteln könnten. Des weiteren weisen die Betroffenen eine deutliche Störung der Baroreflexfunktion - Blutdruckänderungen zu puffern - auf. Wir konnten durch Interphase-FISH (Fluoreszens -in situ- Hybridisation) in mehreren Familien zeigen, dass autosomal-dominante Hypertonie/Brachydaktylie durch chromosomale Umbauten auf dem kurzen Arm von Chromosom 12 verursacht wird (Anita Rauch, Humangenetisches Institut, Universität Erlangen-Nürnberg). Die genaue Kartierung dieser Umbauten, die Klonierung der chromosomalen Bruchpunkte und die Untersuchung von ESTs (expressed sequence tags) in der Region sollten uns ermöglichen, die für die Erkrankung verantwortlichen Gene zu entschlüsseln.
A. Die linke Abbildung zeigt einen Interphasekern eines betroffenen Mitglieds der T�rkischen Familie, der mit Fluoreszenz-markierten BAC-Klonen (Chromosom12p) in der Kombination 1 (96K9 rot-284C17 gr�n-345P1 gelb) hybridisiert wurde. Die Reihenfolge der Klone auf dem rechten Chromosom 12 hat sich im Gegensatz zum linken Chromosm 12, auf dem die Klone in der erwarteten Folge angeordnet sind, ge�ndert. Die rechte Abbildung zeigt einen Zellkern der gleichen Person, hybridisiert mit BAC-Klonen in der Kombination 4 (184C8 gelb-96K9 rot-284C17 gr�n). Auf dem rechte Chromosom ist eine Inversion zwischen den Klonen 184C8 und 96K9 zu sehen.
Hussam Al-Kateb und unsere Gruppe zeigten, dass autosomal-rezessive Hypercholesterinämie in einer Familie aus Syrien auf eine neue Spleißmutation in einem LDL-Rezeptor-Adaptorprotein (ARH) auf Chromosom 1p36.12 zurückzuführen ist. Er und seine Mitarbeiter expremierten diese Mutation in HepG2-Zellen und wiesen nach, dass sie eine Deletion von zwei Nukleotiden auf mRNA-Ebene verursacht und somit zu einem frühzeitigen Abbruch der Proteinsynthese und zu einem verkürzten Adaptorprotein führt.
ausgewählte Publikationen zum Thema:
Bähring S, Rauch A, Toka O, Schroeder C, Hesse C, Siedler H, Fesus G, Haefeli WE, Busjahn A, Aydin A, Neuenfeld Y, Muhl A, Toka HR, Gollasch M, Jordan J, Luft FC. Autosomal-dominant hypertension with type E brachydactyly is caused by rearrangement on the short arm of chromosome 12. Hypertension. 2004;43(2):471-6.
Schuster H, Wienker TE, Bähring S, Bilginturan N, Toka HR, Neitzel H, Jeschke E, Toka O, Gilbert D, Lowe A, Ott J, Haller H, Luft FC. Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat Genet. 1996;13(1):98-100.
Al-Kateb H, Bähring S, Hoffmann K, Strauch K, Busjahn A, Nurnberg G, Jouma M, Bautz EK, Dresel HA, Luft FC. Mutation in the ARH gene and a chromosome 13q locus influence cholesterol levels in a new form of digenic-recessive familial hypercholesterolemia. Circ Res. 2002;90(9):951-8.
Al-Kateb H, Bautz EK, Luft FC, Bähring S. A splice mutation in a Syrian autosomal recessive hypercholesterolemia family causes a two-nucleotide deletion of mRNA. Circ Res. 2003;93(5):e49-50.
Komplexe Erkrankungen
Essentielle (primäre) Hypertonie ist ein entscheidender Risikofaktor für kardiovaskuläre Morbidität und Mortalität. Hoher Blutdruck ist in hohem Maße vererbbar. Die genetischen Faktoren, die zu diesem polygenen (komplexen) Phänotyp beitragen, sind jedoch bis heute weitestgehend unbekannt. Eine große Chinesische Familie und eine Gruppe ausgewählter Chinesischer Patienten und Kernfamilien mit essentieller Hypertonie wurde in der MDC-Arbeitsgruppe von Norbert Hübner, mit der unsere Arbeitsgruppe kooperiert, untersucht. Eine Genom-weite parametrische Kopplungsanalyse führte zur Identifizierung eines neuen Genorts für primäre Hypertonie auf Chromosom 12p (Parametrischer LOD-Score 3,44). Diese chromosomale Region überlappt mit dem Genort für monogene Hypertonie/Brachydaktylie, die bisher einzige monogene Hypertonieform, die eine Ähnlichkeit zu essentieller Hypertonie aufweist. Diese Daten deuten darauf hin, dass diese Region von hoher Relevanz sein sollte, was die Aufklärung neuer Mechanismen der Entstehung von essentieller Hypertonie betrifft.
Um Gene, die am Fettstoffwechsel beteiligt sind, genauer untersuchen zu können, betrieben wir an der Franz-Volhard-Klinik ein �Genetic Field working�-Programm, das auf die Rekrutierung von Drei-Generationen-Familien ausgerichtet war. Unsere Kohorte besteht aus mehr als 250 dieser Familien (1054 Personen). Wir haben für den Lipidmetabolismus relevante und untereinander vernetzte Gene untersucht. Eine umfassende SNP Analyse dieser Gene erlaubte uns die Beteiligung der verschiedenen Genorte am Gesamtgeschehen des Fettstoffwechsels zu studieren. Die Analysen der Daten wurden von Hans Knoblauch in Zusammenarbeit mit Prof. Jens Reich, dem ehemaligen Leiter der Bioinformatik am MDC, durchgeführt.
Die häufigste Todesursache weltweit sind Herz-Kreislauf-Erkrankungen. Cirka die Hälfte der Herztoden sterben an plötzlichem Herztod. Das Long-QT-Syndrom (LQTS) ist klinisch charakterisiert durch eine Verlängerung des im EGK gemessenen QT-Intervalls und damit verbunden mit einem hohen Risiko, einem plötzlichen Herztod zu erliegen, der auf die Entwicklung von ventrikulären Tachyarrhytmien, vetrikuläre Fibrillation und Herzstillstand zurückzuführen ist. Das LQTS ist eine Erbkrankheit, bedingt durch genetisch deteminierte Defekte in den Untereinheiten von die Membran durchspannenden Ionenkanälen. Seltene Mutationen, die bei LQTS-Patienten nachgewiesen wurden, führen zur Verlängerung des QT-Intervalls. Aber auch Personen ohne Mutation in den LQTS-Genen können als Reaktion auf Elektrolytstörungen oder auf bestimmte Medikamente eine Verlängerung der QT-Zeiten aufweisen, die ebenfalls eine Ausprägung maligner Formen ventrikulärer Tachycardie zur Folge haben können. Die Neigung zu derartigen Entwicklungen wird durch genetische Varianz beeinflußt. Atakan Aydin und unsere Gruppe haben fünf bekannte Long-QT-Gene in gesunden Probanden auf genetische Varianten (Single Nucleotide Polymorphisms-SNPs) untersucht und fanden dabei 35 häufige SNPs, was die Vermutung nahe legt, dass es sinnvoll ist, diese Gene auch in der allgemeinen Bevölkerung hinsichtlich ihrer Varianz zu untersuchen. Das Ergebnis der Studie demonstriert sehr eindrucksvoll, dass häufige Varianten in Krankheitsgenen auch in der gesunden Allgemeinbevölkerung die Dauer des QT-Intervalls beeinflussen.
Ein Mitglied unserer Gruppe, Andreas Busjahn, gründete unlängst HealthTwiSt und ist seit 1994 für ein Zwillingsprogramm verantwortlich, das einen wesentlichen Anteil unserer Forschung ausmacht. Mit Hilfe dieser erfolgreichen Studie konnte von einer Reihe von Genorten für die Regulation von Blutdruck, von den LQTS-Genen, von relevanten Genorten des Lipidstoffwechsels Kopplung zu den entsprechenden physiologischen Parametern nachgewiesen werden.
ausgewählte Publikationen zum Thema:
Gong M, Zhang H, Schulz H, Lee YA, Sun K, Bähring S, Luft FC, Nurnberg P, Reis A, Rohde K, Ganten D, Hui R, Hubner N. Genome-wide linkage reveals a locus for human essential (primary) hypertension on chromosome 12p. Hum Mol Genet. 2003;12(11):1273-7.
Aydin A, Bähring S, Dahm S, Guenther UP, Uhlmann R, Busjahn A, Luft FC. Single nucleotide polymorphism map of five long-QT genes. J Mol Med. 2004 Dec 15; [Epub ahead of print]
Busjahn A, Knoblauch H, Faulhaber H-D, Uhlmann R, Hoehe M, Schuster H, Luft FC (1999) The QT interval is linked to two long-QT syndrome loci in normal subjects. Circulation 99:3161�3164
Aydin A, Luft FC, Bähring S. Validation of fluorescence-labeled artificial nonhuman sequences for single-strand conformation polymorphism mutation detection in familial hypercholesterolemia. Anal Biochem. 2004;324(1):16-21.
Angewandte Methoden:
- Präparation genomischer DNA
- PCR (Polymerase-Ketten-Reaktion)
- Genotypisierung von Mikrosatelliten und SNPs
- Physikalische Kartierung von Genen
- Sequenzierung von Plasmiden und BAC/PAC-Enden
- Mutationsanalyse (direkte Sequenzierung, SSCP mittels Kapillarsequenzierautomat)
- Pulse-Field-Gelelektrophorese
- Southernblot
- Zellkultur (Lymphoblastoide Zelllinien, primäre Fibroblasten)
Sitz der AG:
Max-Delbrück-Centrum für Molekulare Medizin Berlin-Buch
Walter-Friedrich-Haus
Robert-Rössle-Str. 10
13125 Berlin
Ständige Mitarbeiter der Arbeitsgruppe:
| Gruppenleiter: |
| Dr. Sylvia Bähring |
Telefon: |
+49-30-9406-2505
|
| Fax: |
+49-30-9406-2536
|
| email: |
[email protected]
|
Wissenschaftler:
Dr. Maolian Gong
Dr. Martin Kann
Dr. Atakan Aydin
Technische Mitarbeiter:
Astrid Mühl
Regina Uhlmann
Yvette Neuenfeld |
